
La dernière publication de votre équipe concerne l’albinisme, un sujet que vous connaissez bien…
Pr Benoît Arveiler : L’albinisme est une anomalie de la pigmentation cutanée et oculaire. Cette maladie rare d’origine génétique touche une personne sur 17000. Nous connaissons actuellement 20 gènes responsables de cette maladie, qui est cliniquement et génétiquement hétérogène. Notre équipe a découvert les 2 derniers gènes.
Nous travaillons sur cette maladie depuis une vingtaine d’années et avons mis en place le diagnostic moléculaire au niveau hospitalier. Plus de 2000 patients ont déjà été analysés, constituant la plus grande cohorte mondiale de patients présentant un albinisme. Ceci constitue un atout pour pouvoir réaliser des études telles que celle que nous avons réalisée avec l’équipe de Manchester.
Quel était l’objectif de cette étude ?
Dr Eulalie Lasseaux : Nous réalisons le diagnostic moléculaire par séquençage nouvelle génération des 20 gènes impliqués dans l’albinisme, ainsi que de gènes de diagnostic différentiel.
Comme l’a dit Benoît, il y a une hétérogénéité clinique et génétique dans cette pathologie, le diagnostic moléculaire est donc indispensable afin d’adapter la prise en charge et le conseil génétique chez les patients.
Dans notre cohorte, nous avons trouvé la cause moléculaire pour 72% des patients. 28% des patients restent donc cependant sans diagnostic moléculaire établi.
Le gène principal d’albinisme est le gène TYR codant la tyrosinase, impliqué dans 40% des cas. Pour la moitié de ces cas, et plus particulièrement chez des patients présentant une forme modérée d’albinisme, le variant hypomorphe R402Q est impliqué. Il s’agit d’un variant fréquent dans la population générale (environ 20%). L’albinisme est une maladie récessive, c'est-à-dire qu'il faut que les deux allèles soient altérés pour que la maladie s’exprime. Dans les maladies rares, classiquement les variants responsables sont très rares dans la population générale. Ce qui est nouveau ici, c’est le fait qu’un variant fréquent (R402Q) associé à un variant rare soit impliqué dans le diagnostic.
Nous avons donc voulu aller plus loin pour mieux comprendre dans quelles conditions ce variant fréquent était impliqué.
Dans cette étude, notre équipe a fourni 1208 patients pour lesquels le diagnostic moléculaire était établi, et l’équipe de Manchester en a fourni 105.
Une cohorte contrôle de 30 000 personnes sans albinisme issue du projet UK Biobank a été inclue dans ce travail. Les 30 000 individus ont eu un séquençage de génome entier, une mesure de l’acuité visuelle et un examen ophtalmologique par Tomographie à Cohérence Optique visant à évaluer l’état de leur fovéa.
Qu’avez-vous découvert ?
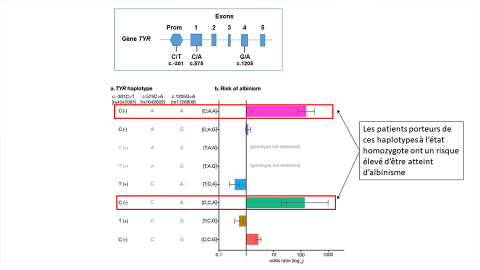
Dr Vincent Michaud : Nous avons étudié le variant hypomorphe R402Q, de même qu’un autre variant fréquent du gène TYR, S192Y, et un variant, fréquent lui aussi, situé dans le promoteur et dont l’effet sur l’expression du gène TYR avait déjà été montré. Nous nous sommes plus particulièrement intéressés aux haplotypes formés par ces trois variants, et avons montré que deux haplotypes, C-A-A et C-C-A, conféraient un risque important d’être atteint d’albinisme en comparaison à tous les autres haplotypes possibles. Ces haplotypes ont une fréquence d’environ 1% dans la population générale, et ont le même effet qu’un variant pathogène rare. En d’autres termes, lorsque l’un ou l’autre de ces haplotypes est présent soit à l’état homozygote soit associé à l’état hétérozygote composite avec un autre variant pathogène de TYR, la maladie apparait.
Quel impact cette découverte va-t-elle avoir sur les patients ?
Dr Vincent Michaud : Cette découverte invite à remodeler le diagnostic moléculaire de l’albinisme. Notamment, la prise en compte de ce nouvel haplotype qui est considéré aussi fort qu’un variant pathogène, augmente le taux de diagnostic positif de 19%, fournissant donc un diagnostic à autant de patients.
Nous nous sommes aussi demandé s’il n’existait pas un haplotype protecteur de l’albinisme et nous avons découvert que certaines combinaisons de ces haplotypes, T-C-A et T-C-G, permettent de protéger de l’albinisme.
Quelles sont les perspectives que cela ouvre ?
Pr Benoît Arveiler : Tout d’abord comme nous l’avons vu ces résultats améliorent le diagnostic des patients. Nous sommes en train de rédiger avec nos collègues de Manchester un article indiquant comment intégrer ces haplotypes dans l’arbre décisionnel du diagnostic de l’albinisme.
Plus largement ces résultats proposent un nouveau paradigme pour l’architecture génétique des maladies rares mendéliennes d’une manière générale : au cas de figure classique d’un allèle pathogène constitué par un variant fort et très rare dans la population, il faut ajouter celui d’une combinaison de variants faibles et fréquents sous la forme d’un haplotype (ou d’un nombre restreint d’haplotypes) pathogène et finalement rare dans la population.
Ce paradigme pourrait expliquer pour partie ce que l’on appelle l’« hérédité manquante » dans bon nombre de maladies génétiques rares.
Dr Vincent Michaud : Ce nouveau paradigme nous incite d’ores et déjà à nous interroger sur l’existence de mécanismes similaires au sein des autres gènes d’albinisme.